Systémový lupus erythematosus (SLE)
Systémový lupus erythematosus (SLE) je chronické autoimunitní onemocnění charakterizované multisystémovým zánětem, sekundární vaskulitidou a produkcí autoprotilátek. Nemoc postihuje především ženy v reprodukčním věku, ale může se vyskytnout i u dětí a mužů. Přibližně 10 % pacientů má pozitivní rodinnou anamnézu.
Epidemiologie
SLE je v dětském věku vzácným onemocněním s roční incidencí mezi 0,3–2,5 na 100 000 dětí do 18 let a prevalencí 1,1-9,7 na 100 000 dětí, u dospělých je pak incidence 1-25 na 100 000. Onemocnění je častější u afroamerické a asijské populace. Poměr dívek ku chlapcům je v dětském věku 5:1, u dospělých žen a mužů až 9:1.
Etiologie a patogeneze
SLE vzniká na základě genetické predispozice, hormonálních vlivů a environmentálních faktorů, které vedou k narušení imunologické tolerance.
Diagnostika
Diagnóza SLE se opírá o klinické projevy a laboratorní testy. Klasifikační kritéria zahrnují kombinaci klinických a imunologických příznaků. Pro diagnózu musí pacient splnit minimálně 4 kritéria, z toho jedno klinické a jedno imunologické, nebo mít histologicky potvrzenou lupusovou nefritidu při přítomnosti autoprotilátek (antinukleárních nebo proti dvouvláknové DNA) (viz tabulka Klasifikační kritéria pro SLE: SLICC 2012).
Klinické projevy
Klinické projevy SLE mohou být od mírných po závažné, zpočátku jsou často nenápadné. Typické je intermitentní postižení různých systémů v různé časové posloupnosti v průběhu měsíců i let.
- Nespecifické projevy: často v začátku onemocnění, subfebrilie až febrilie, hubnutí, nechutenství, únava, generalizovaná lymfadenopatie.
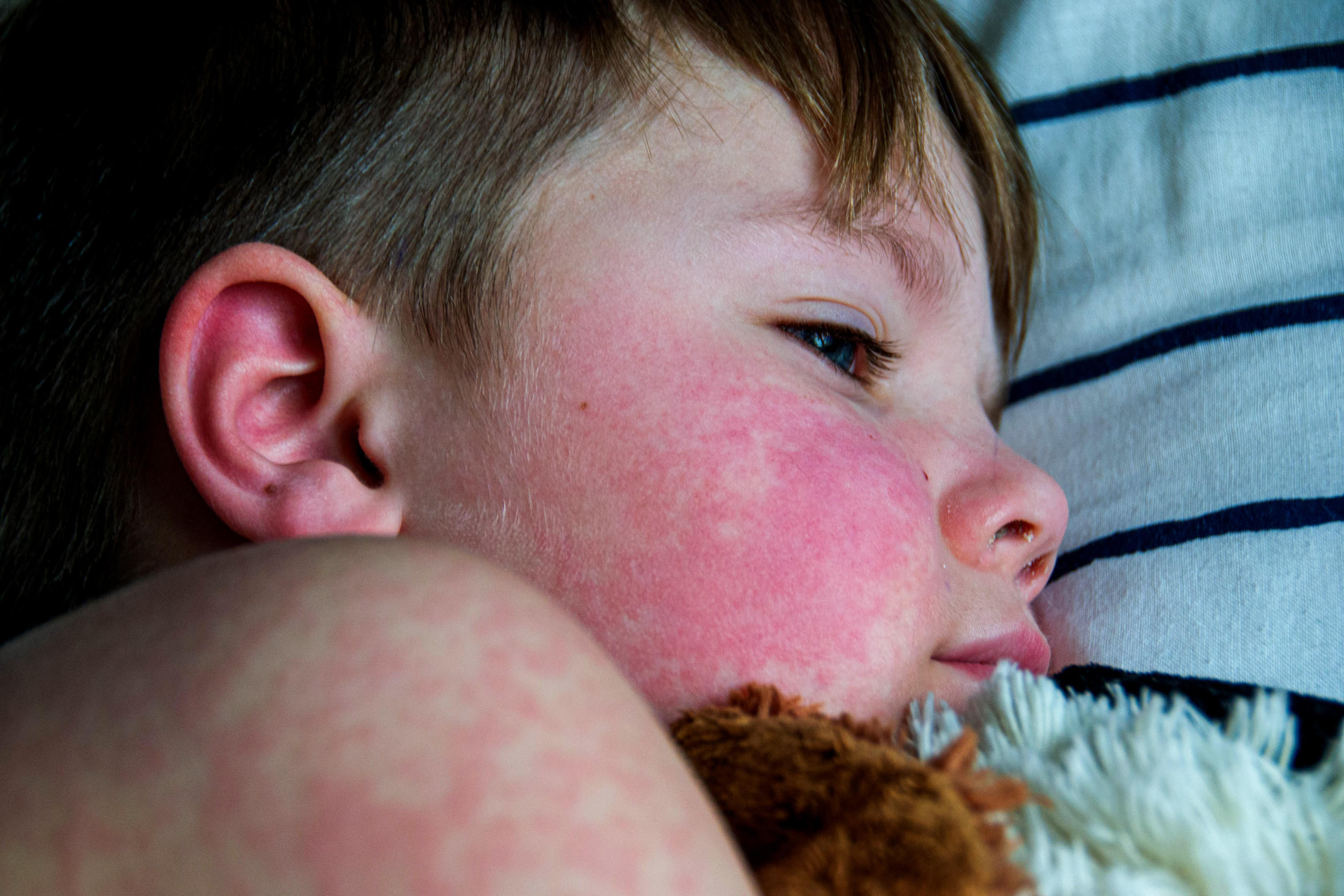
- Kožní: Motýlovitý exantém (image), fotosenzitivita (image), ulcerace, diskoidní vyrážka, lupusová panikulitida, chilblain lupus (image)
- Slizniční: orální (image) a nazální ulcerace
- Muskuloskeletální: Symetrická neerozivní polyartritida, myalgie, proximální svalová slabost, aseptické kostní nekrózy.
- Renální: Lupusová nefritida (proteinurie, hematurie, erytrocytární válce).
- Neuropsychiatrické: Postihují 20-50% dětí. Bolesti hlavy, křeče, akutní stav zmatenosti, neuropatie, myelitida, dyskinetické poruchy (chorea, tremor, parkinsonismus), deprese. Normální nález na MR CNS nevylučuje neuropsychiatrický lupus.
- Hematologické: Hemolytická anémie, leukopenie, trombocytopenie, zvýšený sklon k žilním i arteriálním trombózám a tromboembolickým komplikacím při přítomnosti antifosfolipidových protilátek
- Kardiovaskulární: Perikarditida, myokarditida, infarkt myokardu.
- Plicní: pleuritida, akutní a chronická pneumonitida, plicní hypertenze, akutní plicní hemoragie až apoplexie akutně ohrožující život
- Gastrointestinální: Výrazné bolesti břicha mohou být projevem život ohrožující komplikace: peritonitidy, vaskulitidy, pankreatitidy. Bolestmi břicha s průjmem a hypalbuminémií se může projevit protein-loosing enteropatie.
- Oční: Retinální vaskulitida, subretinální edém, optická neuritida.
- Endokrinopatie: Autoimunitní tyreoiditida, steroidní diabetes, opoždění pohlavního vývoje.
- Syndrom aktivace makrofágů
| SLICC 2012 – klasifikační kritéria pro systémový lupus erythematosusPro diagnózu SLE musí být přítomno minimálně 1 kritérium z klinických a 1 kritérium z imunologických, dohromady minimálně 4 kritéria, nebo biopsií potvrzená lupusová nefritida s pozitivitou ANA nebo anti-dsDNA | |
| Klinická kritéria | Imunologická kritéria |
| Akutní kožní lupus | ANA |
| Chronický kožní lupus | Anti-DNA |
| Orální nebo nazální ulcerace | Anti-SM |
| Nejizvící alopecie | Antifosfolipidové protilátky |
| Artritida | Nízký komplement (C3, C4, CH50) |
| Serozitida | Pozitivní přímý Coombsův test |
| Renální postižení | |
| Neurologické postižení | |
| Hemolytická anémie | |
| Leukopenie | |
| Trombocytopenie | |
Laboratorní diagnostika
- Základní vyšetření: Krevní obraz (anémie, leukopenie, trombocytopenie), FW (zvýšená), CRP (obvykle normální, při zvýšení nutno zvážit infekci), ledvinné a jaterní funkce, močový sediment (proteinurie, hematurie, snížení glomerulární filtrace).
- Imunologické testy: ANA (pozitivní u většiny pacientů), anti-dsDNA (specifické pro SLE), anti-Sm (specifické, ale málo citlivé), antifosfolipidové protilátky (při podezření na antifosfolipidový syndrom (APS), snížené komplementy (C3, C4, CH50).
Diferenciální diagnostika
Stavy kdy pomýšlet na SLE:
- U dívek v pubertálním věku s artritidou, teplotami, fotosenzitivním exantémem ,
- V případě klinického obrazu idiopatické trombocytopenické purpury (ITP) s pozitivitou ANA,
- U nejasné žilní trombózy
- V případě chorey či jiného dyskinetického syndromu,
- Při nálezu hyperbilirubinemie u dospívající dívky pomýšlet na možnost hemolytické anemie při SLE.
Léčba
Nefarmakologická
- Ochrana před UV zářením.
- Prevence infekcí (imunizace, důsledná léčba)
- Omezení stresu, dostatek odpočinku, pestrá strava.
Farmakologická
- Hydroxychlorochin: Základní léčba u všech pacientů, protizánětlivý a vaskuloprotektivní účinek.
- Imunosupresivní léčba: kortikosteroidy, azathioprin, mykofenolát mofetil, cyklofosfamid, B-buňky depletující léčba
- Symptomatická
- Antikoagulace: U antifosfolipidového syndromu.
- ACE inhibitory: renoprotektivní léčba při proteinurii
- Pomocná: např. prevence glukokortikoidy indukované osteoporózy
Prognóza
SLE je chronické onemocnění s relabujícím průběhem. Hlavními faktory ovlivňujícími prognózu jsou lupusová nefritida, případně infekční komplikace, z dlouhodobého hlediska pak kardiovaskulární komplikace. Včasná diagnóza a adekvátní léčba výrazně zlepšují prognózu a kvalitu života pacientů.
Juvenilní dermatomyozitida
Úvod
Juvenilní dermatomyozitida (JDM) je vzácné autoimunitní onemocnění neznámé etiologie. K rozvoji JDM přispívají genetické faktory, infekční spouštěče a UV záření. Přibližně 70 % pacientů má autoprotilátky specifické pro myozitidu (MSA), jako jsou anti-NXP2 nebo anti-TIF-1γ, nebo protilátky asociované s myozitidou (MAA), které se vyskytují i u jiných systémových onemocnění.
Klinický obraz
Diagnóza JDM je založena na kombinaci klinických kritérií, laboratorních a pomocných vyšetření, přičemž neexistuje žádný specifický laboratorní test.
Systémové projevy Až 60 % pacientů trpí celkovými příznaky, jako je únava, slabost, horečka, nechutenství, pokles hmotnosti a změny chování (podrážděnost, deprese).
Kožní projevy jsou přítomny u 100 % pacientů, v 73–100 % případů již při první manifestaci onemocnění. Mezi typické léze patří heliotropní exantém (fialové zbarvení očních víček – image), malární vyrážka přesahující kořen nosu, Gottronovy papuly na extenzorových plochách kloubů (image) a teleangiektázie na očních víčkách. U těžkých forem mohou vznikat ulcerace.
Postižení svalů Postižení příčně pruhovaných svalů se vyskytuje u 80–100 % pacientů (chybí u amyopatické formy JDM). Typická je symetrická slabost proximálních svalů horních i dolních končetin, flexorů krku a břišních svalů. Časté jsou obtíže při vstávání (Gowersův příznak), únava a neobratnost.
Postižení dalších orgánů
- Klouby: Symetrická polyartritida (6–42 % pacientů), ranní ztuhlost, kontraktury.
- Dystrofická kalcinóza: Ukládání vápníku v podkoží a fasciích, často spojené s autoprotilátkami anti-NXP2. (image)
- Gastrointestinální trakt: Riziko ischémie střevní stěny a perforace, ulcerace sliznic dutiny ústní.
- Plíce: Intersticiální pneumopatie s rizikem plicní hypertenze.
- Myokard: Srdeční postižení je u dětí vzácné.
Diagnostika
Diagnóza JDM se opírá o klinické a laboratorní projevy. Nová klasifikační kritéria (Klasifikační kritéria pro Idiopatické zánětlivé myopatie: EULAR/ACR, 2017) stratifikují pacienty do 3 skupin dle pravděpodobnosti onemocnění. Skóre lze jednoduše spočítat prostřednictvím online kalkulačky: IIM calculator – English version.
Laboratorní vyšetření
- Zánětlivá aktivita: Obvykle nebývá výrazně zvýšená – FW a CRP mohou být normální nebo mírně zvýšené.
- Krevní obraz: Obvykle v normě, ale může se objevit leukopenie, Coombs pozitivní anémie či trombocytopenie (zejména při překryvném syndromu se systémovým lupusem).
- Imunofenotypizace: Zvýšený počet CD20+ B-lymfocytů.
- Biochemie: Zvýšené svalové enzymy (ALT, AST, LD, CK), zvýšený myoglobin, zvýšený von Willebrandův faktor (marker vaskulopatie).
- Imunologie: Pozitivní antinukleární protilátky (ANA), protilátky specifické pro myozitidu (anti-Jo1, anti-Mi2, anti-SRP, anti-NXP2, anti-MDA5) a protilátky asociované s jinými systémovými onemocněními (anti-dsDNA, anti-Sm, anti-SSA, anti-SSB, anti-U1-RNP). Některé protilátky korelují s konkrétními klinickými projevy, jako je plicní postižení nebo kalcinóza.
Zobrazovací a funkční vyšetření
- Hodnocení svalové síly: Pomocí standardizovaných testů, jako jsou CMAS (Childhood Myositis Assessment Scale) a MMT8 (Manual Muscle Test).
- Magnetická rezonance svalů: Umožňuje zobrazit zánětlivé změny. MR se využívá k monitorování efektu léčby a k výběru vhodného svalu pro biopsii.
- Kapilaroskopie nehtových lůžek: Odhaluje redukci počtu kapilár, jejich strukturální změny a změny v uspořádání (dezorganizaci). Nález koreluje s aktivitou onemocnění.
- Kardiologické vyšetření: EKG, echokardiografie a RTG hrudníku mohou pomoci při podezření na srdeční nebo plicní postižení.
- Biopsie svalu: Provádí se cíleně ze svalu s největším postižením dle MR, pomáhá při stanovení prognózy.
- Funkční vyšetření plic: Spirometrie a difuzní kapacita plic odhalují případné respirační komplikace.
- CT vyšetření plic (HRCT): Objektivizace přítomnosti a rozsahu intersticiálních plicních změn
Diferenciální diagnostika
Důležité je odlišit JDM od jiných onemocnění s podobnými příznaky:
- Kožní onemocnění: Psoriáza (odlišná mikroangiopatie při vyšetření nehtového lůžka).
- Neuromuskulární choroby: Duchennova a Beckerova muskulární dystrofie a další hereditární myopatie, metabolické myopatie (Pompeho choroba, poruchy β-oxidace mastných kyselin).
- Revmatologická onemocnění: Juvenilní idiopatická artritida, systémová sklerodermie, systémový lupus erythematosus, smíšené onemocnění pojiva.
- Endokrinopatie: Hypotyreóza, hypertyreóza.
- Infekční myozitidy: Viry (chřipka, coxsackie), bakteriální pyomyozitida, trichinelóza.
Stavy kdy pomýšlet na JDM
- U každého dítěte s proximální svalovou slabostí a minimálním kožním nálezem
- U dětí s typickým kožním nálezem s i bez svalové slabosti
- U dětí se zvýšením markerů svalového postižení bez zjevné svalové slabosti s minimálním nebo žádným kožním nálezem
Léčba
Nefarmakologická
- Ochrana před UV zářením.
- Rehabilitace zaměřená na odstranění kontraktur a posílení svalstva.
- Prevence aspirace při poruše polykání.
Farmakologická
- Imunosupresivní léčba: kortikosteroidy, methotrexát, cyklosporin A
- Imunomodulační léčba: intravenózní imunoglobuliny (IVIG)
- Biologická léčba: Rezervována pro rezistentní případy (blokátory TNF-α, anti-CD20).
Prognóza
Díky včasné agresivní imunosupresivní léčbě se mortalita JDM snížila na 1–2 %. Přesto je dlouhodobá morbidita vysoká a pouze 30 % pacientů dosáhne remise bez nutnosti další léčby. Výskyt kardiomyopatie a intersticiální pneumopatie je nižší než u dospělých pacientů s dermatomyozitidou. Na rozdíl od dospělých JDM nesouvisí s malignitami. Včasná diagnostika a zahájení terapie jsou klíčové pro prognózu pacientů s JDM.
Text připravil Prokop Šeferna (prokop.seferna@vfn.cz), převážně na základě knihy Dětská revmatologie v praxi, kapitoly 18 Idiopatické zápalové myopatie (Tomáš Dallos, dallos@nudch.eu) a 20 Juvenilní systémový lupus erythematosus (Hana Malcová – Hana.Malcova@fnmotol.cz, Veronika Vargová – veronika.vargova@upjs.sk).
Více příspěvků pro zdravotnické odborníky




