Systemic lupus erythematosus (SLE)
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease characterized by multisystem inflammation, secondary vasculitis, and the production of autoantibodies. The disease primarily affects women of reproductive age, but it can also occur in children and men. Approximately 10% of patients have a positive family history.
Epidemiology
SLE is a rare disease in childhood, with an annual incidence of 0.3–2.5 per 100,000 children under 18 years of age and a prevalence of 1.1–9.7 per 100,000 children. In adults, the incidence is 1–25 per 100,000. The disease is more common in African American and Asian populations. The female-to-male ratio is 5:1 in childhood and up to 9:1 in adults.
Etiology and Pathogenesis
SLE develops on the basis of genetic predisposition, hormonal influences, and environmental factors that lead to a disruption of immunological tolerance.
Diagnosis
The diagnosis of SLE is based on clinical manifestations and laboratory tests. The classification criteria include a combination of clinical and immunological features. For diagnosis, the patient must meet at least four criteria, including at least one clinical and one immunological, or have histologically confirmed lupus nephritis in the presence of autoantibodies (antinuclear or anti–double-stranded DNA) (see table Classification Criteria for SLE: SLICC 2012).
Clinical Manifestations
The clinical manifestations of SLE may range from mild to severe and are often subtle at the onset. A characteristic feature is the intermittent involvement of various organ systems in different chronological sequences over months or even years.
- Nonspecific manifestations: Often present at the onset of disease; low-grade fever to high fever, weight loss, loss of appetite, fatigue, generalized lymphadenopathy.
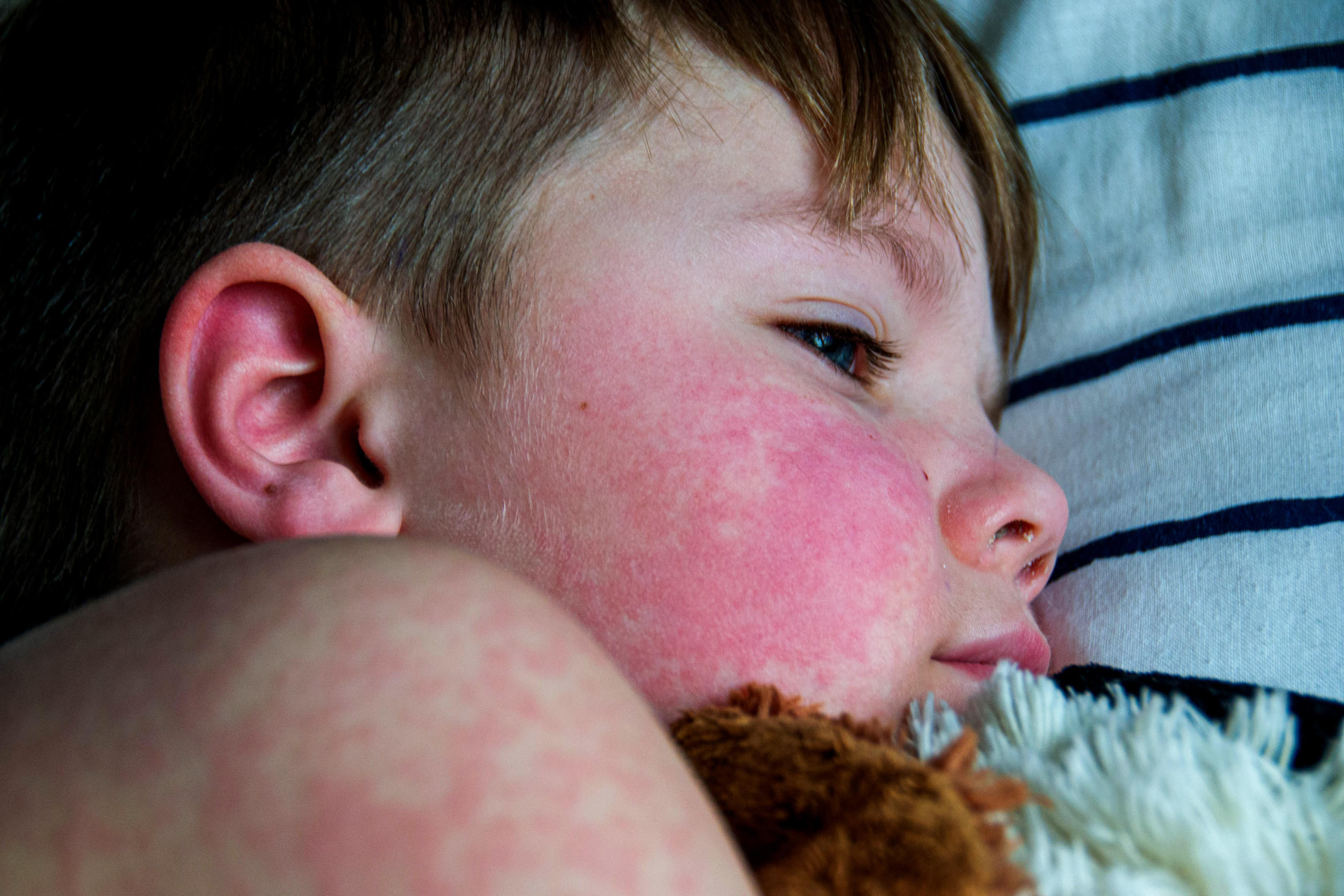
- Cutaneous: Malar (“butterfly”) rash (image), photosensitivity (image), ulcerations, discoid rash, lupus panniculitis, chilblain lupus (image)
- Mucosal: Oral (image) and nasal ulcerations
- Musculoskeletal: Symmetrical non-erosive polyarthritis, myalgia, proximal muscle weakness, aseptic bone necrosis.
- Renal: Lupus nephritis (proteinuria, hematuria, erythrocyte casts).
- Neuropsychiatric: Affect 20–50% of children. Headaches, seizures, acute confusional states, neuropathies, myelitis, dyskinetic disorders (chorea, tremor, parkinsonism), depression. A normal CNS MRI does not exclude neuropsychiatric lupus.
- Hematologic: Hemolytic anemia, leukopenia, thrombocytopenia, increased risk of venous and arterial thromboses and thromboembolic complications in the presence of antiphospholipid antibodies.
- Cardiovascular: Pericarditis, myocarditis, myocardial infarction.
- Pulmonary: Pleuritis, acute and chronic pneumonitis, pulmonary hypertension, acute pulmonary hemorrhage up to life-threatening apoplexy.
- Gastrointestinal: Severe abdominal pain may be a manifestation of life-threatening complications such as peritonitis, vasculitis, or pancreatitis. Abdominal pain with diarrhea and hypoalbuminemia may indicate protein-losing enteropathy.
- Ocular: Retinal vasculitis, subretinal edema, optic neuritis.
- Endocrinopathies: Autoimmune thyroiditis, steroid-induced diabetes, delayed pubertal development.
- Macrophage Activation Syndrome
| SLICC 2012 – Classification Criteria for Systemic Lupus ErythematosusFor the diagnosis of SLE, at least one clinical criterion and one immunological criterion must be present, with a total of at least four criteria, or biopsy-proven lupus nephritis in the presence of positive ANA or anti–dsDNA antibodies. | |
| Clinical Criteria | Immunological Criteria |
| Acute cutaneous lupus | ANA |
| Chronic cutaneous lupus | Anti-DNA |
| Oral or nasal ulcerations | Anti-SM |
| Non-scarring alopecia | Antiphospholipid antibodies |
| Arthritis | Low complement (C3, C4, CH50) |
| Serositis | Positive direct Coombs test |
| Renal involvement | |
| Neurological involvement | |
| Hemolytic anemia | |
| Leukopenia | |
| Thrombocytopenia | |
Laboratory Diagnostics
- Basic tests: Complete blood count (anemia, leukopenia, thrombocytopenia), ESR (elevated), CRP (usually normal; if elevated, infection should be considered), renal and liver function tests, urinalysis (proteinuria, hematuria, decreased glomerular filtration).
- Immunological tests: ANA (positive in most patients), anti-dsDNA (specific for SLE), anti-Sm (specific but with low sensitivity), antiphospholipid antibodies (in suspected antiphospholipid syndrome, APS), decreased complement levels (C3, C4, CH50).
Differential Diagnosis
Situations in which SLE should be considered:
- In adolescent girls with arthritis, fever, and photosensitive rash,
- In the case of idiopathic thrombocytopenic purpura (ITP) with positive ANA,
- In cases of unexplained venous thrombosis
- In patients with chorea or another dyskinetic syndrome,
- In adolescent girls with hyperbilirubinemia, consider the possibility of hemolytic anemia associated with SLE.
Treatment
Non-pharmacological
- Protection from UV radiation.
- Infection prevention (immunization, adequate treatment)
- Stress reduction, sufficient rest, balanced diet.
Pharmacological
- Hydroxychloroquine: Basic treatment for all patients, with anti-inflammatory and vasculoprotective effects.
- Immunosuppressive therapy: Corticosteroids, azathioprine, mycophenolate mofetil, cyclophosphamide, B-cell depleting therapy.
- Symptomatic treatment
- Anticoagulation: In antiphospholipid syndrome.
- ACE inhibitors: Renoprotective treatment in proteinuria
- Supportive: e.g., prevention of glucocorticoid-induced osteoporosis
Prognosis
SLE is a chronic disease with a relapsing course. The main factors influencing prognosis are lupus nephritis and, potentially, infectious complications; in the long term, cardiovascular complications also play a key role. Early diagnosis and adequate treatment significantly improve prognosis and quality of life for patients.
Juvenile Dermatomyositis
Introduction
Juvenile dermatomyositis (JDM) is a rare autoimmune disease of unknown etiology. The development of JDM is influenced by genetic factors, infectious triggers, and UV radiation. Approximately 70% of patients have myositis-specific autoantibodies (MSA), such as anti-NXP2 or anti-TIF-1γ, or myositis-associated autoantibodies (MAA), which are also found in other systemic diseases.
Clinical Picture
The diagnosis of JDM is based on a combination of clinical criteria, laboratory findings, and ancillary investigations; there is no single specific laboratory test.
Systemic manifestations Up to 60% of patients present with general symptoms such as fatigue, weakness, fever, loss of appetite, weight loss, and behavioral changes (irritability, depression).
Cutaneous manifestations Present in 100% of patients, in 73–100% of cases already at the first manifestation of the disease. Typical lesions include heliotrope rash (purple discoloration of the eyelids – image), malar rash extending across the nasal bridge, Gottron’s papules on the extensor surfaces of the joints (image) and telangiectasias on the eyelids. Ulcerations may occur in severe forms.
Muscle involvement Involvement of striated muscles occurs in 80–100% of patients (absent in the amyopathic form of JDM). The typical feature is symmetrical weakness of the proximal muscles of the upper and lower limbs, neck flexors, and abdominal muscles. Common difficulties include trouble rising from a seated position (Gowers’ sign), fatigue, and clumsiness.
Involvement of Other Organs
- Joints: Symmetrical polyarthritis (6–42% of patients), morning stiffness, contractures.
- Dystrophic calcinosis: Calcium deposition in subcutaneous tissue and fascia, often associated with anti-NXP2 autoantibodies. (image)
- Gastrointestinal tract: Risk of intestinal wall ischemia and perforation, ulcerations of the oral mucosa.
- Lungs: Interstitial pneumonitis with a risk of pulmonary hypertension.
- Myocardium: Cardiac involvement is rare in children.
Diagnosis
The diagnosis of JDM is based on clinical and laboratory manifestations. The new classification criteria (Classification Criteria for Idiopathic Inflammatory Myopathies: EULAR/ACR, 2017) stratify patients into three groups according to the probability of disease. The score can easily be calculated using an online calculator: IIM calculator – English version.
Laboratory Investigations
- Inflammatory activity: Usually not markedly elevated – ESR and CRP may be normal or only mildly increased.
- Complete blood count: Usually within normal limits, but leukopenia, Coombs-positive anemia, or thrombocytopenia may occur (especially in overlap syndrome with systemic lupus).
- Immunophenotyping: Increased number of CD20+ B lymphocytes.
- Biochemistry: Elevated muscle enzymes (ALT, AST, LD, CK), increased myoglobin, elevated von Willebrand factor (marker of vasculopathy).
- Immunology: Positive antinuclear antibodies (ANA), myositis-specific antibodies (anti-Jo1, anti-Mi2, anti-SRP, anti-NXP2, anti-MDA5), and myositis-associated antibodies (anti-dsDNA, anti-Sm, anti-SSA, anti-SSB, anti-U1-RNP). Some antibodies correlate with specific clinical manifestations, such as pulmonary involvement or calcinosis.
Imaging and Functional Investigations
- Assessment of muscle strength: Using standardized tests such as CMAS (Childhood Myositis Assessment Scale) and MMT8 (Manual Muscle Test).
- Magnetic resonance imaging (MRI) of muscles: Allows visualization of inflammatory changes. MRI is used to monitor treatment response and to select the most appropriate muscle for biopsy.
- Nailfold capillaroscopy: Reveals a reduction in the number of capillaries, their structural alterations, and disorganization. The findings correlate with disease activity.
- Cardiological evaluation: ECG, echocardiography, and chest X-ray may assist in suspected cardiac or pulmonary involvement.
- Muscle biopsy: Performed selectively from the muscle most affected according to MRI; useful in determining prognosis.
- Pulmonary function tests: Spirometry and diffusion capacity of the lungs help detect possible respiratory complications.
- CT scan of the lungs (HRCT): Provides objective evaluation of the presence and extent of interstitial lung disease.
Differential Diagnosis
It is important to distinguish JDM from other diseases with similar symptoms:
- Skin diseases: Psoriasis (different microangiopathy on nailfold examination).
- Neuromuscular disorders: Duchenne and Becker muscular dystrophy and other hereditary myopathies, metabolic myopathies (Pompe disease, disorders of β-oxidation of fatty acids).
- Rheumatologic diseases: Juvenile idiopathic arthritis, systemic scleroderma, systemic lupus erythematosus, mixed connective tissue disease.
- Endocrinopathies: Hypothyroidism, hyperthyroidism.
- Infectious myositis: Viruses (influenza, coxsackie), bacterial pyomyositis, trichinellosis.
Situations in which JDM should be considered
- In every child with proximal muscle weakness and minimal skin findings
- In children with typical skin findings with or without muscle weakness
- In children with elevated markers of muscle involvement without obvious muscle weakness and with minimal or no skin manifestations
Treatment
Non-pharmacological
- Protection from UV radiation.
- Rehabilitation aimed at eliminating contractures and strengthening muscles.
- Prevention of aspiration in swallowing disorders.
Pharmacological
- Immunosuppressive therapy: Corticosteroids, methotrexate, cyclosporine A
- Immunomodulatory therapy: Intravenous immunoglobulins (IVIG)
- Biological therapy: Reserved for resistant cases (TNF-α blockers, anti-CD20).
Prognosis
Thanks to early and aggressive immunosuppressive treatment, the mortality of JDM has decreased to 1–2%. Nevertheless, long-term morbidity remains high, and only 30% of patients achieve remission without the need for further therapy. The incidence of cardiomyopathy and interstitial pneumopathy is lower than in adult patients with dermatomyositis. Unlike adults, JDM is not associated with malignancies. Early diagnosis and initiation of therapy are crucial for the prognosis of patients with JDM.
Text prepared by Prokop Šeferna (prokop.seferna@vfn.cz), mainly based on Dětská revmatologie v praxi, chapters 18 Idiopatické zápalové myopatie (Tomáš Dallos, dallos@nudch.eu) and 20 Juvenilní systémový lupus erythematosus (Hana Malcová – Hana.Malcova@fnmotol.cz, Veronika Vargová – veronika.vargova@upjs.sk).
More posts for medical professionals




