Lupus eritematos sistemic (LES)
Lupusul eritematos sistemic (LES) este o boală autoimună cronică, caracterizată prin inflamație multisistemică, vasculită secundară și producerea de autoanticorpi. Boala afectează în principal femeile aflate la vârsta reproductivă, dar poate apărea și la copii și bărbați. Aproximativ 10% dintre pacienți au antecedente familiale pozitive.
Epidemiologie
LES este o boală rară în copilărie, cu o incidență anuală de 0,3–2,5 la 100.000 de copii sub 18 ani și o prevalență de 1,1–9,7 la 100.000. La adulți, incidența este de 1–25 la 100.000. Boala este mai frecventă în populațiile afro-americane și asiatice. Raportul femei/bărbați este de 5:1 în copilărie și poate ajunge până la 9:1 la adulți.
Etiologie și patogeneză
LES se dezvoltă pe baza unei predispoziții genetice, a influențelor hormonale și a factorilor de mediu care duc la perturbarea toleranței imunologice.
Diagnostic
Diagnosticul LES se bazează pe manifestările clinice și pe testele de laborator. Criteriile de clasificare includ o combinație de trăsături clinice și imunologice. Pentru stabilirea diagnosticului, pacientul trebuie să îndeplinească cel puțin patru criterii, dintre care cel puțin unul clinic și unul imunologic, sau să aibă nefrită lupică confirmată histologic în prezența autoanticorpilor (antinucleari sau anti–dublu lanț ADN) (vezi tabelul Criterii de clasificare pentru LES: SLICC 2012).
Manifestări clinice
Manifestările clinice ale LES pot varia de la forme ușoare la severe și sunt adesea subtile la debut. O caracteristică distinctivă este afectarea intermitentă a diferitelor sisteme de organe, în succesiuni cronologice variabile, de-a lungul lunilor sau chiar anilor.
- Manifestări nespecifice: Frecvent prezente la debutul bolii; febră ușoară până la febră înaltă, scădere în greutate, pierderea apetitului, oboseală, limfadenopatie generalizată.
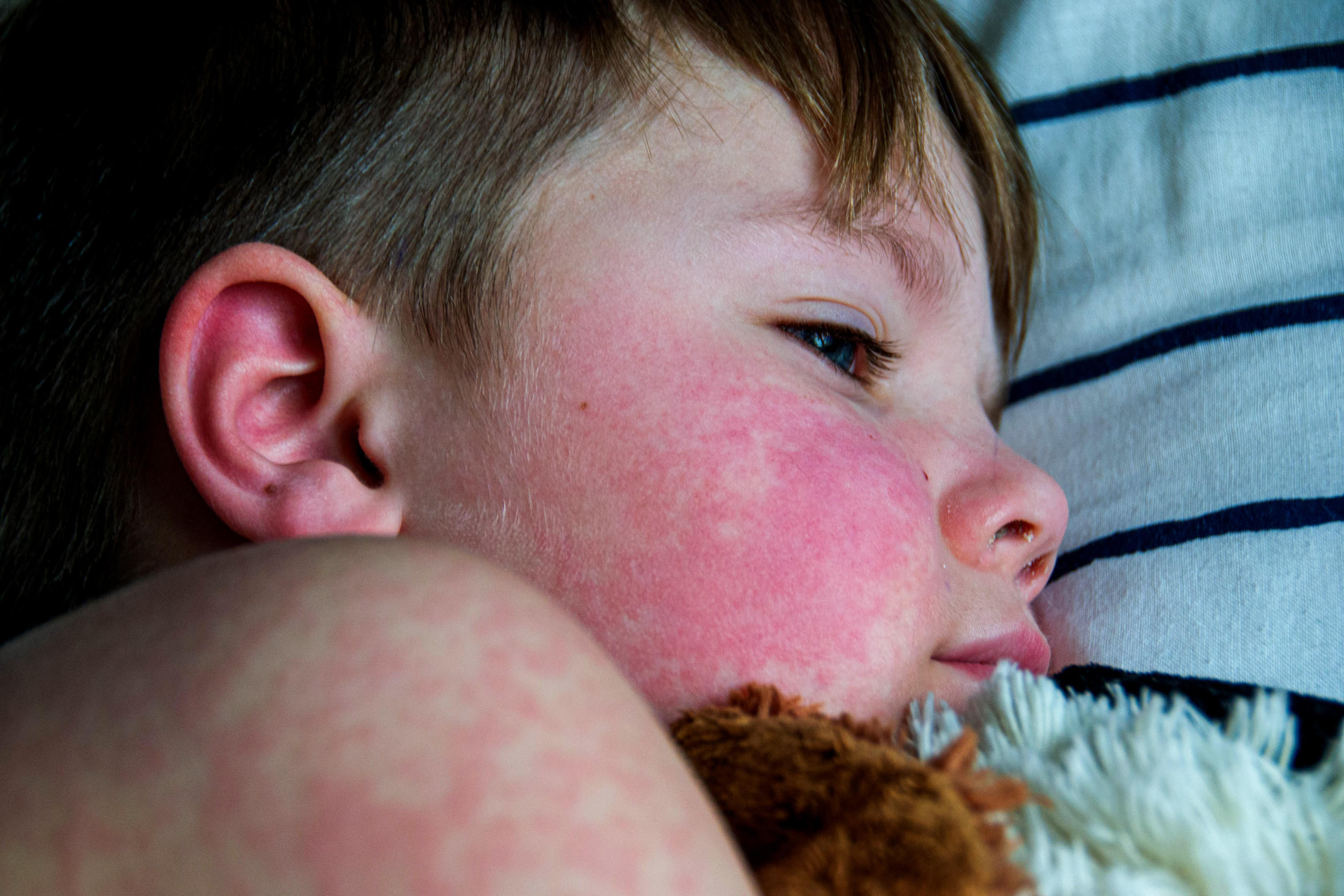
- Cutante: Eritem malar („în fluture”) (image), fotosensibilitate (image), ulcerații, erupție discoidă, paniculită lupică, lupus pernio (image)
- Mucoase: Ulcerații orale (image) și nazale.
- Musculoscheletale: Poliartrită simetrică neerozivă, mialgii, slăbiciune musculară proximală, necroză osoasă aseptică.
- Renale: Nefrită lupică (proteinurie, hematurie, cilindri eritrocitari).
- Neuropsihiatrice: Afectează 20–50% dintre copii. Cefalee, convulsii, stări confuzionale acute, neuropatii, mielită, tulburări diskinetice (coree, tremor, parkinsonism), depresie. Un RMN cerebral normal nu exclude lupusul neuropsihiatric.
- Hematologice: Anemie hemolitică, leucopenie, trombocitopenie, risc crescut de tromboze venoase și arteriale și complicații tromboembolice în prezența anticorpilor antifosfolipidici.
- Cardiovasculare: Pericardită, miocardită, infarct miocardic.
- Pulmonare: Pleurită, pneumonită acută și cronică, hipertensiune pulmonară, hemoragie pulmonară acută până la apoplexie care pune viața în pericol.
- Gastrointestinale: Durerea abdominală severă poate fi o manifestare a unor complicații amenințătoare de viață, cum ar fi peritonita, vasculita sau pancreatita. Durerea abdominală asociată cu diaree și hipoalbuminemie poate indica enteropatie cu pierdere de proteine.
- Oculare: Vasculită retiniană, edem subretinian, nevrită optică.
- Endocrinopatii: Tiroidită autoimună, diabet indus de steroizi, întârziere în dezvoltarea pubertară.
- Sindromul de activare a macrofagelor
| SLICC 2012 – Criterii de clasificare pentru lupusul eritematos sistemicPentru diagnosticul LES, trebuie să fie prezente cel puțin un criteriu clinic și unul imunologic, cu un total de minimum patru criterii, sau nefrită lupică confirmată prin biopsie în prezența anticorpilor ANA pozitivi sau anti–dsDNA. | |
| Criterii clinice | Criterii imunologice |
| Lupus cutanat acut | ANA |
| Lupus cutanat cronic | Anticorpi anti-ADN |
| Ulcerații orale sau nazale | Anticorpi anti-Sm |
| Alopecie fără cicatrici | Anticorpi antifosfolipidici |
| Artrită | Complement scăzut (C3, C4, CH50) |
| Serozită | Test Coombs direct pozitiv |
| Afectare renală | |
| Afectare neurologică | |
| Anemie hemolitică | |
| Leucopenie | |
| Trombocitopenie | |
Diagnostic de laborator
- Teste de bază: Hemoleucogramă completă (anemie, leucopenie, trombocitopenie), VSH (crescut), CRP (de obicei normal; dacă este crescut, trebuie luată în considerare o infecție), teste ale funcției renale și hepatice, sumar de urină (proteinurie, hematurie, scăderea ratei de filtrare glomerulară).
- Teste imunologice: ANA (pozitiv la majoritatea pacienților), anticorpi anti-dsDNA (specifici pentru LES), anticorpi anti-Sm (specifici, dar cu sensibilitate scăzută), anticorpi antifosfolipidici (în caz de suspiciune de sindrom antifosfolipidic, APS), scăderea nivelurilor complementului (C3, C4, CH50).
Diagnostic diferențial
Situații în care trebuie luat în considerare LES:
- La adolescente cu artrită, febră și erupție fotosensibilă,
- În caz de purpură trombocitopenică idiopatică (ITP) cu ANA pozitiv,
- În cazuri de tromboză venoasă de cauză inexplicabilă,
- La pacienți cu coree sau alt sindrom diskinetic,
- La adolescente cu hiperbilirubinemie, trebuie luată în considerare posibilitatea unei anemii hemolitice asociate cu LES.
Tratament
Ne-farmacologic
- Protecție împotriva radiațiilor UV.
- Prevenirea infecțiilor (imunizare, tratament adecvat).
- Reducerea stresului, odihnă suficientă, dietă echilibrată.
Farmacologic
- Hidroxiclorochină: Tratament de bază pentru toți pacienții, cu efecte antiinflamatorii și vasculoprotectoare.
- Terapie imunosupresoare: Corticosteroizi, azatioprină, micofenolat mofetil, ciclofosfamidă, terapie depleționantă a celulelor B.
- Tratament simptomatic
- Anticoagulare: În sindromul antifosfolipidic.
- Inhibitori ai ECA: Tratament nefroprotector în caz de proteinurie.
- Suportiv: de exemplu, prevenirea osteoporozei induse de glucocorticoizi.
Prognostic
LES este o boală cronică, cu evoluție recurentă. Principalii factori care influențează prognosticul sunt nefrita lupică și, potențial, complicațiile infecțioase; pe termen lung, complicațiile cardiovasculare joacă, de asemenea, un rol important. Diagnosticul precoce și tratamentul adecvat îmbunătățesc semnificativ prognosticul și calitatea vieții pacienților.
Dermatomiozită juvenilă
Introducere
Dermatomiozita juvenilă (JDM) este o boală autoimună rară, de etiologie necunoscută. Dezvoltarea JDM este influențată de factori genetici, factori infecțioși declanșatori și radiații UV. Aproximativ 70% dintre pacienți prezintă autoanticorpi specifici pentru miozită (MSA), precum anti-NXP2 sau anti-TIF-1γ, sau autoanticorpi asociați miozitei (MAA), care pot fi întâlniți și în alte boli sistemice.
Tablou clinic
Diagnosticul JDM se bazează pe o combinație de criterii clinice, constatări de laborator și investigații suplimentare; nu există un test de laborator unic, specific bolii.
Manifestări sistemice Până la 60% dintre pacienți prezintă simptome generale precum oboseală, slăbiciune, febră, pierderea apetitului, scădere în greutate și modificări de comportament (irritabilitate, depresie).
Manifestări cutanate Prezente la 100% dintre pacienți, în 73–100% dintre cazuri încă de la prima manifestare a bolii. Leziunile tipice includ erupția heliotropă (decolorare violacee a pleoapelor – image),erupția malară care se extinde peste baza nasului, papulele Gottron pe suprafețele extensoare ale articulațiilor (image) și telangiectaziile pleoapelor. În formele severe pot apărea ulcerații.
Afectarea musculară Afectarea mușchilor striați apare la 80–100% dintre pacienți (absentă în forma amiopatică de JDM). Caracteristica tipică este slăbiciunea simetrică a mușchilor proximali ai membrelor superioare și inferioare, a flexorilor gâtului și a mușchilor abdominali. Dificultățile frecvente includ ridicarea dificilă din poziția șezând (semnul Gowers), oboseală și stângăcie motorie.
Afectarea altor organe
- Articulații: Poliartrită simetrică (6–42% dintre pacienți), rigiditate matinală, contracturi.
- Calcinoză distrofică: Depunerea calciului în țesutul subcutanat și în fascie, adesea asociată cu autoanticorpii anti-NXP2. (image)
- Tract gastrointestinal: Risc de ischemie și perforație a peretelui intestinal, ulcerații ale mucoasei bucale.
- Plămâni: Pneumonită interstițială cu risc de hipertensiune pulmonară.
- Miocard: Afectarea cardiacă este rară la copii.
Diagnostic
Diagnosticul JDM se bazează pe manifestările clinice și de laborator. Noile criterii de clasificare (Criteriile de clasificare pentru miozitele inflamatorii idiopatice: EULAR/ACR, 2017) împart pacienții în trei grupe în funcție de probabilitatea bolii. Scorul poate fi calculat cu ușurință folosind un calculator online: IIM calculator – English version.
Investigații de laborator
- Activitate inflamatorie: De obicei, nu este semnificativ crescută – VSH și CRP pot fi normale sau doar ușor crescute.
- Hemoleucogramă completă: De obicei în limite normale, dar pot apărea leucopenie, anemie Coombs-pozitivă sau trombocitopenie (în special în sindromul de suprapunere cu lupus eritematos sistemic).
- Imunofenotipare: Număr crescut de limfocite B CD20+.
- Biochimie: Creșterea enzimelor musculare (ALT, AST, LD, CK), creșterea mioglobinei, creșterea factorului von Willebrand (marker al vasculopatiei).
- Imunologie: Anticorpi antinucleari (ANA) pozitivi, anticorpi specifici pentru miozită (anti-Jo1, anti-Mi2, anti-SRP, anti-NXP2, anti-MDA5) și anticorpi asociați miozitei (anti-dsDNA, anti-Sm, anti-SSA, anti-SSB, anti-U1-RNP). Unii anticorpi se corelează cu manifestări clinice specifice, cum ar fi afectarea pulmonară sau calcinoza.
Investigații imagistice și funcționale
- Evaluarea forței musculare: Folosind teste standardizate, cum ar fi CMAS (Childhood Myositis Assessment Scale) și MMT8 (Manual Muscle Test).
- Rezonanța magnetică (RMN) a mușchilor: Permite vizualizarea modificărilor inflamatorii. RMN-ul este utilizat pentru monitorizarea răspunsului la tratament și pentru selectarea mușchiului cel mai potrivit pentru biopsie.
- Capilaroscopia pliurilor unghiale: Evidențiază reducerea numărului de capilare, modificările structurale și dezorganizarea acestora. Rezultatul se corelează cu activitatea bolii.
- Evaluare cardiologică: ECG, ecocardiografie și radiografie toracică pot fi utile în caz de suspiciune de afectare cardiacă sau pulmonară.
- Biopsia musculară: Se efectuează selectiv din mușchiul cel mai afectat conform RMN-ului; este utilă în determinarea prognosticului.
- Testele funcționale pulmonare: Spirometria și măsurarea capacității de difuzie pulmonară ajută la depistarea posibilelor complicații respiratorii.
- Tomografia computerizată a plămânilor (HRCT): Oferă o evaluare obiectivă a prezenței și extinderii bolii pulmonare interstițiale.
Diagnostic diferențial
Este important să se facă distincția între JDM și alte boli cu simptome similare:
- Boli cutanate: Psoriazis (microangiopatie diferită la examinarea pliurilor unghiale).
- Afecțiuni neuromusculare: Distrofia musculară Duchenne și Becker și alte miopatii ereditare, miopatii metabolice (boala Pompe, tulburări ale β-oxidării acizilor grași).
- Boli reumatologice: Artrită idiopatică juvenilă, sclerodermie sistemică, lupus eritematos sistemic, boală mixtă de țesut conjunctiv.
- Endocrinopatii: Hipotiroidism, hipertiroidism.
- Miozită infecțioasă: Virusuri (gripă, coxsackie), piomiozită bacteriană, trichineloză.
Situații în care JDM trebuie avută în vedere
- La orice copil cu slăbiciune musculară proximală și leziuni cutanate minime.
- La copii cu leziuni cutanate tipice, cu sau fără slăbiciune musculară.
- La copii cu markeri serici ai afectării musculare crescuți, fără slăbiciune musculară evidentă și cu manifestări cutanate minime sau absente.
Tratament
Ne-farmacologic
- Protecție împotriva radiațiilor UV.
- Reabilitare orientată spre eliminarea contracturilor și întărirea musculaturii.
- Prevenirea aspirației în tulburările de deglutiție.
Farmacologic
- Terapie imunosupresoare: Corticosteroizi, metotrexat, ciclosporină A.
- Terapie imunomodulatoare: Imunoglobuline intravenoase (IVIG).
- Terapie biologică: Rezervată cazurilor rezistente (blocanți TNF-α, anti-CD20).
Prognostic
Datorită tratamentului imunosupresor precoce și agresiv, mortalitatea JDM a scăzut la 1–2%. Cu toate acestea, morbiditatea pe termen lung rămâne ridicată, iar doar 30% dintre pacienți ating remisia fără necesitatea unui tratament suplimentar. Incidența cardiomiopatiei și a pneumopatiei interstițiale este mai scăzută decât la pacienții adulți cu dermatomiozită. Spre deosebire de adulți, JDM nu este asociată cu malignități. Diagnosticul precoce și inițierea timpurie a terapiei sunt esențiale pentru prognosticul pacienților cu JDM.
Text pregătit de Prokop Šeferna (prokop.seferna@vfn.cz), bazat în principal pe Dětská revmatologie v praxi, capitolele 18 Idiopatické zápalové myopatie (Tomáš Dallos, dallos@nudch.eu) și 20 Juvenilní systémový lupus erythematosus (Hana Malcová – Hana.Malcova@fnmotol.cz, Veronika Vargová – veronika.vargova@upjs.sk).
Mai multe postări pentru profesioniștii din domeniul medical




