Системски лупус еритематозус (СЛЕ)
Системски лупус еритематозус (СЛЕ) је хронична аутоимуна болест коју карактерише мултисистемска инфламација, секундарни васкулитис и продукција аутоантитела. Болест углавном погађа жене репродуктивног доба, али се може јавити и код деце и мушкараца. Приближно 10% пацијената има позитивну породичну анамнезу.
Епидемиологија
СЛЕ је ретка болест у детињству, са годишњом инциденцом од 0,3–2,5 на 100.000 деце млађе од 18 година и преваленцом од 1,1–9,7 на 100.000 деце. Код одраслих, инциденца износи 1–25 на 100.000. Болест је чешћа код афроамеричке и азијске популације. Однос жена и мушкараца је 5:1 у детињству и до 9:1 код одраслих.
Етиологија и патогенеза
СЛЕ се развија на основу генетске предиспозиције, хормонских утицаја и фактора из окружења који доводе до поремећаја имунотолеранције.
Дијагноза
Дијагноза СЛЕ се заснива на клиничким манифестацијама и лабораторијским тестовима. Класификациони критеријуми обухватају комбинацију клиничких и имунолошких карактеристика. За постављање дијагнозе, пацијент мора испуњавати најмање четири критеријума, од којих бар један клинички и један имунолошки, или имати хистолошки потврђен лупусни нефритис у присуству аутоантитела (антинуклеарних или анти–двоструколанчане ДНК) (види табелу Класификациони критеријуми за СЛЕ: SLICC 2012).
Клиничке манифестације
Клиничке манифестације СЛЕ могу варирати од благих до тешких и често су суптилне на почетку болести. Карактеристична особина је повремено укључивање различитих органских система у различитим временским секвенцама током месеци или чак година.
- Неспецифичне манифестације: Често присутне на почетку болести; блага до висока температура, губитак телесне тежине, губитак апетита, умор, генерализована лимфаденопатија.
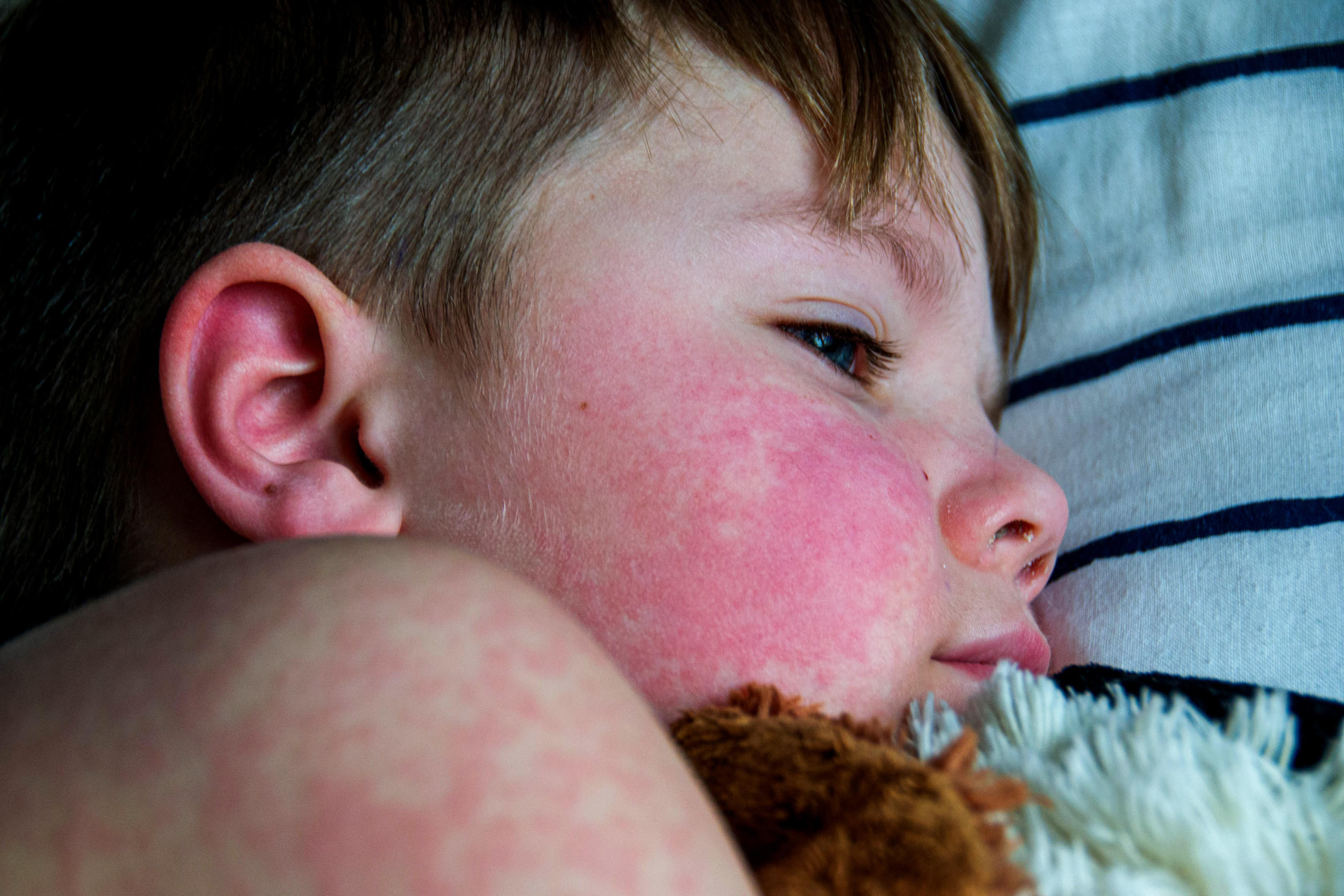
- Кожне: Маларијални („лептир“) осип (image), фотосензитивност (image), улцерације, дискоидни осип, лупус паникулитис, хладни лупус (image)
- Мукозалне: Улцерације у усној дупљи (image) и у носу.
- Мишићно-скелетне: Симетрични, неерозивни полиартритис, миалгија, слабост проксималних мишића, асептична некроза костију.
- Бубрежне: Лупусни нефритис (протеинурија, хематурија, еритроцитни цилиндри).
- Неуропсихијатријске: Погађају 20–50% деце. Главобоље, епилептични напади, акутна конфузна стања, неуропатије, мијелитис, дискинетички поремећаји (хореја, тремор, паркинсонизам), депресија. Нормалан налаз МРИ мозга не искључује неуропсихијатријски лупус.
- Хематолошке: Хемолитичка анемија, леукопенија, тромбоцитопенија, повећан ризик од венских и артеријских тромбоза и тромбоемболијских компликација у присуству антифосфолипидних антитела.
- Кардиоваскуларне: Перикардитис, миокардитис, инфаркт миокарда.
- Плућне: Плеуритис, акутни и хронични пнеумонитис, плућна хипертензија, акутно плућно крварење до животно угрожавајуће апоплексије.
- Гастроинтестиналне: Јак бол у стомаку може бити манифестација потенцијално фаталних компликација као што су перитонитис, васкулитис или панкреатитис. Бол у стомаку са дијарејом и хипоалбуминемијом може указивати на ентеропатију са губитком протеина.
- Офталмолошке: Васкулитис мрежњаче, субретинални едем, оптички неуритис.
- Ендокринопатије: Аутоимуни тироидитис, дијабетес изазван стероидима, одложен пубертетски развој.
- Синдром активације макрофага
| SLICC 2012 – Класификациони критеријуми за системски лупус еритематозусЗа дијагнозу СЛЕ потребно је да буду присутна најмање један клинички и један имунолошки критеријум, са укупно најмање четири критеријума, или биопсијом потврђен лупусни нефритис у присуству позитивних ANA или анти–dsDNA антитела. | |
| Клинички критеријуми | Имунолошки критеријуми |
| Акутни кожни лупус | ANA |
| Хронични кожни лупус | Анти-ДНК антитела |
| Улцерације у устима или носу | Анти-СМ антитела |
| Небраздајућа алопеција | Антифосфолипидна антитела |
| Артритис | Снижен комплемент (C3, C4, CH50) |
| Серозитис | Позитиван директни Кумбсов тест |
| Бубрежно оштећење | |
| Неуролошко оштећење | |
| Хемолитичка анемија | |
| Леукопенија | |
| Тромбоцитопенија | |
Лабораторијска дијагностика
- Основни тестови: Комплетна крвна слика (анемија, леукопенија, тромбоцитопенија), ЕСР (повишен), ЦРП (обично нормалан; ако је повишен, треба размотрити инфекцију), тестови функције бубрега и јетре, анализа урина (протеинурија, хематурија, смањена гломеруларна филтрација).
- Имунолошки тестови: ANA (позитиван код већине пацијената), анти-dsDNA (специфичан за СЛЕ), анти-Sm (специфичан али с ниском осетљивошћу), антифосфолипидна антитела (у случају сумње на антифосфолипидни синдром, APS), смањен ниво комплемента (C3, C4, CH50).
Диференцијална дијагноза
Ситуације у којима треба размотрити СЛЕ:
- Код адолесцентних девојака са артритисом, грозницом и фотосензитивним осипом,
- У случају идиопатске тромбоцитопеничне пурпуре (ИТП) са позитивним ANA,
- Код необјашњене венске тромбозе,
- Код пацијената са хорејом или другим дискинетичким синдромом,
- Код адолесцентних девојака са хипербилирубинемијом треба размотрити могућност хемолитичке анемије повезане са СЛЕ.
Терапија
Нефармаколошка
- Заштита од УВ зрачења.
- Превенција инфекција (имунизација, адекватно лечење).
- Смањење стреса, довољно одмора, уравнотежена исхрана.
Фармаколошка
- Хидроксихлорокин: Основни третман за све пацијенте, са антиинфламаторним и васкулопротективним ефектом.
- Имуносупресивна терапија: Кортикостероиди, азатиоприн, микофенолат мофетил, циклофосфамид, терапија усмерена на деактивацију B-ћелија.
- Симптоматска терапија
- Антикоагулантна терапија: Код антифосфолипидног синдрома.
- ACE инхибитори: Нефропротективни третман у случају протеинурије.
- Потпорна терапија: нпр. превенција остеопорозе изазване глукокортикоидима.
Прогноза
СЛЕ је хронична болест са рецидивирајућим током. Главни фактори који утичу на прогнозу су лупусни нефритис и, потенцијално, инфективне компликације; на дужи рок, кардиоваскуларне компликације такође имају кључну улогу. Рана дијагноза и адекватна терапија значајно побољшавају прогнозу и квалитет живота пацијената.
Јувенилни дерматомиозитис
Увод
Јувенилни дерматомиозитис (ЈДМ) је ретка аутоимуна болест непознате етиологије. Развој ЈДМ-а је под утицајем генетских фактора, инфективних окидача и УВ зрачења. Приближно 70% пацијената има аутоантитела специфична за миозитис (МСА), као што су анти-NXP2 или анти-TIF-1γ, или аутоантитела повезана са миозитисом (МАА), која се јављају и код других системских болести.
Клиничка слика
Дијагноза ЈДМ заснива се на комбинацији клиничких критеријума, лабораторијских налаза и додатних испитивања; не постоји један специфичан лабораторијски тест.
Системске манифестације До 60% пацијената има опште симптоме као што су умор, слабост, грозница, губитак апетита, губитак телесне тежине и промене у понашању (раздражљивост, депресија).
Кожне манифестације Присутне су код 100% пацијената, у 73–100% случајева већ при првој манифестацији болести. Типичне лезије укључују хелиотропни осип (љубичаста промена боје капака – image), маларијални осип који се шири преко носног корена, Готронове папуле на екстензорним површинама зглобова (image) и телеангиектазије на капцима. Код тежих облика могу се јавити улцерације.
Захваћеност мишића Захваћеност пругастих мишића јавља се код 80–100% пацијената (одсутна код амиопатичног облика ЈДМ). Типична је симетрична слабост проксималних мишића горњих и доњих екстремитета, флексора врата и трбушних мишића. Чести проблеми укључују потешкоће при устајању из седећег положаја (Говерсов знак), умор и неспретност у покретима.
Захваћеност других органа
- Зглобови: Симетрични полиартритис (6–42% пацијената), јутарња укоченост, контрактуре.
- Дистрофична калциноза: Таложење калцијума у поткожном ткиву и фасцијама, често повезано са анти-NXP2 аутоантителима. (image)
- Гастроинтестинални тракт: Ризик од исхемије и перфорације цревног зида, улцерације оралне слузокоже.
- Плућа: Интерстицијални пнеумонитис са ризиком од плућне хипертензије.
- Миокард: Срчано захваћање је ретко код деце.
Дијагноза
Дијагноза ЈДМ заснива се на клиничким и лабораторијским манифестацијама. Нови класификациони критеријуми (Класификациони критеријуми за идиопатске инфламаторне миопатије: EULAR/ACR, 2017) сврставају пацијенте у три групе према вероватноћи болести. Резултат се лако може израчунати помоћу онлајн калкулатора: IIM calculator – English version.
Лабораторијска испитивања
- Инфламаторна активност: Обично није изразито повећана – ЕСР и ЦРП могу бити нормални или благо повишени.
- Комплетна крвна слика: Обично у границама нормале, али се може јавити леукопенија, Кумбс-позитивна анемија или тромбоцитопенија (посебно код синдрома преклапања са системским лупусом).
- Имунофенотипизација: Повећан број CD20+ B лимфоцита.
- Биохемија: Повишени мишићни ензими (АЛТ, АСТ, ЛД, ЦК), повећан миоглобин, повишен фактор фон Вилебранда (маркер васкулопатије).
- Иммунологија: Позитивна антинуклеарна антитела (ANA), антитела специфична за миозитис (anti-Jo1, anti-Mi2, anti-SRP, anti-NXP2, anti-MDA5) и антитела повезана са миозитисом (anti-dsDNA, anti-Sm, anti-SSA, anti-SSB, anti-U1-RNP). Нека антитела корелирају са одређеним клиничким манифестацијама, као што су захваћеност плућа или калциноза.
Снимања и функционална испитивања
- Процена мишићне снаге: Користе се стандардизовани тестови као што су CMAS (Childhood Myositis Assessment Scale) и MMT8 (Manual Muscle Test).
- Магнетна резонанца (МРИ) мишића: Омогућава визуализацију инфламаторних промена. МРИ се користи за праћење одговора на терапију и за одабир најпогоднијег мишића за биопсију.
- Капилароскопија нокатних набора: Открива смањење броја капилара, њихове структурне промене и дезорганизацију. Налаз корелира са активношћу болести.
- Кардиолошка процена: ЕКГ, ехокардиографија и рендген грудног коша могу помоћи у случају сумње на срчано или плућно захваћање.
- Биопсија мишића: Ради се селективно из мишића који је највише захваћен према налазу МРИ; корисна је за одређивање прогнозе.
- Функционална испитивања плућ: Спирометрија и мерење дифузионог капацитета плућа помажу у откривању могућих респираторних компликација.
- ЦТ плућа (HRCT): Омогућава објективну процену присуства и обима интерстицијалне болести плућа.
Диференцијална дијагноза
Важно је разликовати ЈДМ од других болести са сличним симптомима:
- Кожне болести: Псоријаза (различита микроангиопатија при прегледу нокатних набора).
- Неуромускуларни поремећаји: Дишенова и Бекерова мишићна дистрофија и друге наследне миопатије, метаболичке миопатије (Помпеова болест, поремећаји β-оксидације масних киселина).
- Реуматолошке болести: Јувенилни идиопатски артритис, системска склеродермија, системски лупус еритематозус, мешана болест везивног ткива.
- Ендокринопатије: Хипотироидизам, хипертироидизам.
- Инфективни миозитис: Вируси (грипа, коксакје), бактеријски пиомиозитис, трихинелоза.
Ситуације у којима треба размотрити ЈДМ
- Код сваког детета са проксималном мишићном слабиношћу и минималним кожним променама.
- Код деце са типичним кожним променама, са или без мишићне слабости.
- Код деце са повишеним маркерима мишићног оштећења без изражене мишићне слабости и са минималним или одсутним кожним променама.
Терапија
Нефармаколошка
- Заштита од УВ зрачења.
- Рехабилитација усмерена на уклањање контрактура и јачање мишића.
- Превенција аспирације код поремећаја гутања.
Фармаколошка
- Имуносупресивна терапија: Кортикостероиди, метотрексат, циклоспорин А.
- Имуномодулаторна терапија: Интравенски имуноглобулини (IVIG).
- Биолошка терапија: Резервисана за резистентне случајеве (TNF-α блокатори, анти-CD20).
Прогноза
Захваљујући раној и агресивној имуносупресивној терапији, морталитет код ЈДМ смањен је на 1–2%. Ипак, дугорочна морбидитет остаје висок, а само 30% пацијената постиже ремисију без потребе за додатним лечењем. Учесталост кардиомиопатије и интерстицијалне пнеумопатије је мања него код одраслих пацијената са дерматомиозитисом. За разлику од одраслих, ЈДМ није повезан са малигнитетима. Рана дијагноза и благовремено започињање терапије од кључног су значаја за прогнозу пацијената са ЈДМ.
Текст припремио Прокоп Шеферна (prokop.seferna@vfn.cz), углавном на основу књиге Dětská revmatologie v praxi, поглавља 18 Idiopatické zápalové myopatie (Томаш Даллос, dallos@nudch.eu) и 20 Juvenilní systémový lupus erythematosus (Хана Малцова – Hana.Malcova@fnmotol.cz, Вероника Варгова – veronika.vargova@upjs.sk).
Још објава За здравствене раднике




