Toczeń rumieniowaty układowy (SLE)
Toczeń rumieniowaty układowy (SLE) to przewlekła choroba autoimmunologiczna charakteryzująca się wieloukładowym stanem zapalnym, wtórnym zapaleniem naczyń oraz wytwarzaniem autoprzeciwciał. Choroba dotyczy głównie kobiet w wieku rozrodczym, ale może występować również u dzieci i mężczyzn. Około 10% pacjentów ma dodatni wywiad rodzinny.
Epidemiologia
SLE jest rzadką chorobą w wieku dziecięcym — roczna zapadalność wynosi 0,3–2,5 na 100 000 dzieci poniżej 18 roku życia, a częstość występowania 1,1–9,7 na 100 000. U dorosłych zapadalność wynosi 1–25 na 100 000. Choroba częściej występuje w populacjach afroamerykańskich i azjatyckich. Stosunek kobiet do mężczyzn wynosi 5:1 wśród dzieci i aż 9:1 u dorosłych.
Etiologia i patogeneza
SLE rozwija się na podłożu predyspozycji genetycznych, wpływów hormonalnych oraz czynników środowiskowych, które prowadzą do zaburzenia tolerancji immunologicznej.
Rozpoznanie
Rozpoznanie SLE opiera się na objawach klinicznych oraz badaniach laboratoryjnych. Kryteria klasyfikacyjne obejmują kombinację cech klinicznych i immunologicznych. Do postawienia diagnozy pacjent musi spełniać co najmniej cztery kryteria, w tym co najmniej jedno kliniczne i jedno immunologiczne, lub mieć histologicznie potwierdzone toczniowe zapalenie nerek w obecności autoprzeciwciał (przeciwjądrowych lub przeciw dwuniciowemu DNA) (zob. tabela Kryteria klasyfikacyjne dla SLE: SLICC 2012).
Objawy kliniczne
Objawy kliniczne SLE mogą mieć przebieg od łagodnego do ciężkiego i często są subtelne na początku choroby. Charakterystyczną cechą jest naprzemienne zajmowanie różnych układów narządowych w odmiennych sekwencjach czasowych, trwających miesiące lub nawet lata.
- Objawy nieswoiste: Często obecne na początku choroby; stan podgorączkowy do wysokiej gorączki, utrata masy ciała, brak apetytu, zmęczenie, uogólniona limfadenopatia.
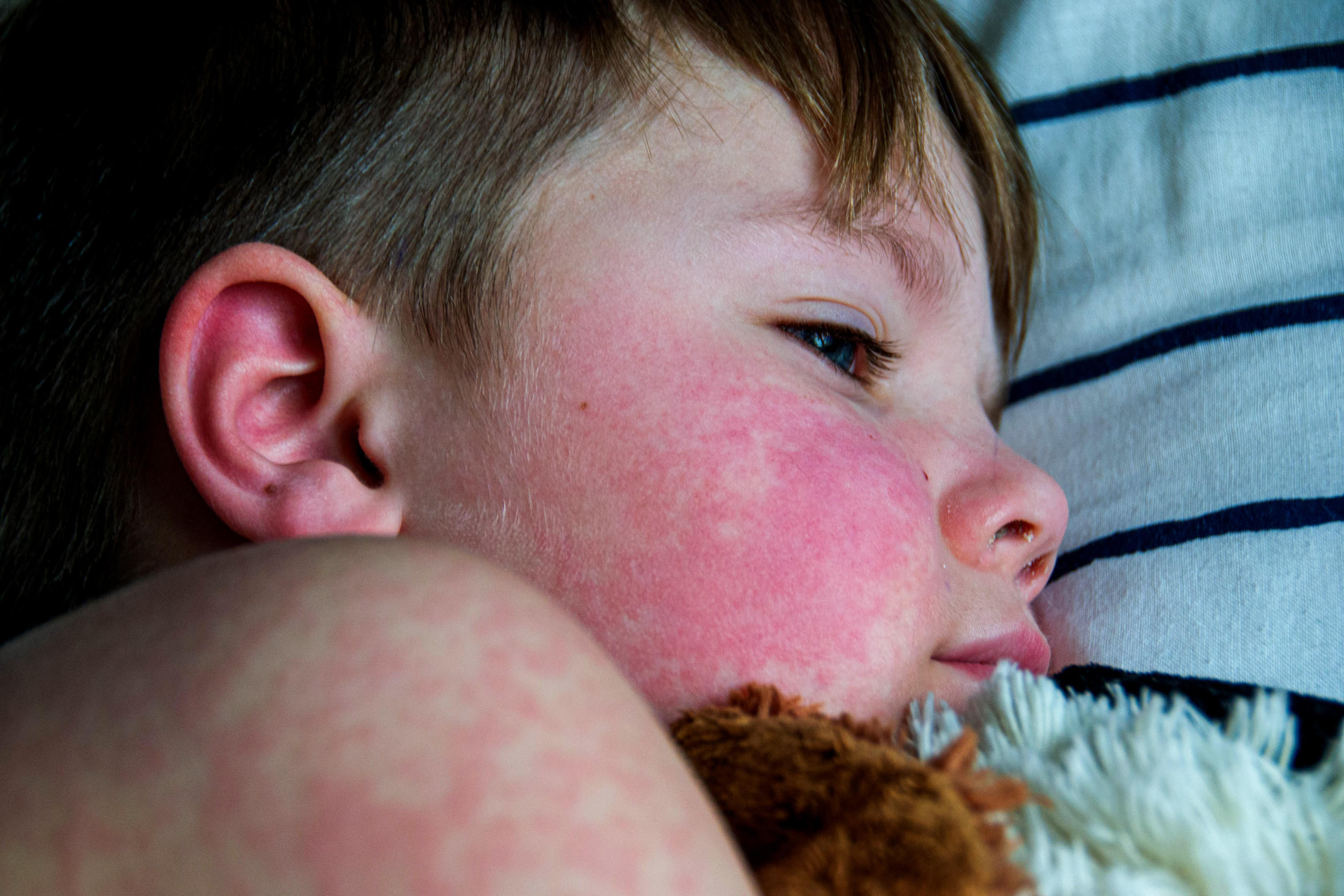
- Skórne: Rumień motylkowaty (malar, image), nadwrażliwość na światło (image), owrzodzenia, wysypka krążkowa, zapalenie tkanki podskórnej (lupus panniculitis), odmrozinowy toczeń (image).
- Śluzówkowe: Owrzodzenia jamy ustnej (image) i nosa.
- Mięśniowo-szkieletowe: Symetryczne, nieerozyjne zapalenie stawów (polyartritis), bóle mięśni (myalgia), osłabienie mięśni proksymalnych, jałowa martwica kości.
- Nerkowe: Toczniowe zapalenie nerek (proteinuria, krwiomocz, wałeczki erytrocytowe).
- Neuropsychiatryczne: Dotyczą 20–50% dzieci. Bóle głowy, napady padaczkowe, ostre stany splątania, neuropatie, zapalenie rdzenia kręgowego, zaburzenia ruchowe (chorea, drżenie, parkinsonizm), depresja. Prawidłowy wynik MRI ośrodkowego układu nerwowego nie wyklucza tocznia neuropsychiatrycznego.
- Hematologiczne: Niedokrwistość hemolityczna, leukopenia, trombocytopenia, zwiększone ryzyko zakrzepicy żylnej i tętniczej oraz powikłań zakrzepowo-zatorowych w obecności przeciwciał antyfosfolipidowych.
- Sercowo-naczyniowe: Zapalenie osierdzia, zapalenie mięśnia sercowego, zawał mięśnia sercowego.
- Płucne: Zapalenie opłucnej, ostre i przewlekłe zapalenie płuc, nadciśnienie płucne, ostry krwotok płucny aż do zagrażającego życiu udaru krwotocznego.
- Żołądkowo-jelitowe: Silny ból brzucha może być objawem zagrażających życiu powikłań, takich jak zapalenie otrzewnej, zapalenie naczyń lub zapalenie trzustki. Ból brzucha z biegunką i hipoalbuminemią może wskazywać na enteropatię z utratą białka.
- Oczne: Zapalenie naczyń siatkówki, obrzęk podsiatkówkowy, zapalenie nerwu wzrokowego.
- Endokrynopatie: Autoimmunologiczne zapalenie tarczycy, cukrzyca indukowana steroidami, opóźnione dojrzewanie płciowe.
- Zespół aktywacji makrofagów
| SLICC 2012 – Kryteria klasyfikacyjne dla tocznia rumieniowatego układowego (SLE)Do rozpoznania SLE wymagane jest spełnienie co najmniej jednego kryterium klinicznego i jednego kryterium immunologicznego, łącznie co najmniej czterech kryteriów, lub histologicznie potwierdzone toczniowe zapalenie nerek w obecności dodatnich przeciwciał ANA lub przeciw dsDNA. | |
| Kryteria kliniczne | Kryteria immunologiczne |
| Ostry skórny toczeń rumieniowaty | ANA |
| Przewlekły skórny toczeń rumieniowaty | Przeciwciała anty-DNA |
| Owrzodzenia jamy ustnej lub nosa | Przeciwciała anty-Sm |
| Niebliznowaciejące łysienie | Przeciwciała antyfosfolipidowe |
| Zapalenie stawów | Obniżony poziom dopełniacza (C3, C4, CH50) |
| Zapalenie błon surowiczych | Dodatni bezpośredni test Coombsa |
| Zajęcie nerek | |
| Zajęcie układu nerwowego | |
| Niedokrwistość hemolityczna | |
| Leukopenia | |
| Trombocytopenia | |
Diagnostyka laboratoryjna
- Podstawowe badania: Morfologia krwi (niedokrwistość, leukopenia, trombocytopenia), OB (podwyższone), CRP (zazwyczaj prawidłowe; jeśli podwyższone, należy rozważyć zakażenie), badania czynności nerek i wątroby, badanie ogólne moczu (białkomocz, krwiomocz, obniżony współczynnik filtracji kłębuszkowej).
- Badania immunologiczne: ANA (dodatnie u większości pacjentów), przeciwciała anty-dsDNA (specyficzne dla SLE), przeciwciała anty-Sm (specyficzne, ale o niskiej czułości), przeciwciała antyfosfolipidowe (w podejrzeniu zespołu antyfosfolipidowego – APS), obniżony poziom dopełniacza (C3, C4, CH50).
Diagnostyka różnicowa
Sytuacje, w których należy rozważyć SLE:
- U dorastających dziewcząt z zapaleniem stawów, gorączką i nadwrażliwością na światło,
- W przypadku idiopatycznej plamicy małopłytkowej (ITP) z dodatnim wynikiem ANA,
- W przypadkach niewyjaśnionej zakrzepicy żylnej,
- U pacjentów z pląsawicą lub innym zespołem dyskinetycznym,
- U dorastających dziewcząt z hiperbilirubinemią należy rozważyć możliwość niedokrwistości hemolitycznej związanej z SLE.
Leczenie
Niefarmakologiczne
- Ochrona przed promieniowaniem UV.
- Profilaktyka zakażeń (szczepienia, odpowiednie leczenie).
- Redukcja stresu, odpowiednia ilość odpoczynku, zrównoważona dieta.
Farmakologiczne
- Hydroksychlorochina: Leczenie podstawowe u wszystkich pacjentów, o działaniu przeciwzapalnym i wazkuloprotekcyjnym.
- Terapia immunosupresyjna: Kortykosteroidy, azatiopryna, mykofenolan mofetylu, cyklofosfamid, leczenie ukierunkowane na eliminację komórek B.
- Leczenie objawowe
- Antykoagulacja: W zespole antyfosfolipidowym.
- Inhibitory ACE: Leczenie nefroprotekcyjne w przypadku białkomoczu.
- Leczenie wspomagające: np. profilaktyka osteoporozy indukowanej glikokortykosteroidami.
Rokowanie
SLE jest chorobą przewlekłą o nawrotowym przebiegu. Głównymi czynnikami wpływającymi na rokowanie są toczeniowe zapalenie nerek oraz ewentualne powikłania infekcyjne; w dłuższej perspektywie kluczową rolę odgrywają również powikłania sercowo-naczyniowe. Wczesne rozpoznanie i odpowiednie leczenie znacząco poprawiają rokowanie oraz jakość życia pacjentów.
Młodzieńcza dermatomiozytis (JDM)
Wprowadzenie
Młodzieńcza dermatomiozytis (JDM) to rzadka choroba autoimmunologiczna o nieznanej etiologii. Na jej rozwój wpływają czynniki genetyczne, infekcyjne czynniki wyzwalające oraz promieniowanie UV. Około 70% pacjentów ma autoprzeciwciała swoiste dla zapalenia mięśni (MSA), takie jak przeciwciała anti-NXP2 lub anti-TIF-1γ, bądź przeciwciała związane z zapaleniem mięśni (MAA), które mogą występować również w innych chorobach układowych.
Obraz kliniczny
Rozpoznanie JDM opiera się na połączeniu kryteriów klinicznych, wyników badań laboratoryjnych oraz badań pomocniczych — nie istnieje pojedynczy, swoisty test laboratoryjny.
Objawy ogólnoustrojowe U nawet 60% pacjentów występują objawy ogólne, takie jak zmęczenie, osłabienie, gorączka, utrata apetytu, spadek masy ciała oraz zmiany w zachowaniu (drażliwość, depresja).
Objawy skórne Występują u 100% pacjentów, w 73–100% przypadków już przy pierwszej manifestacji choroby. Typowe zmiany to rumień heliotropowy (fioletowe zabarwienie powiek – image), rumień motylkowaty obejmujący nasadę nosa, grudki Gottrona na powierzchniach wyprostnych stawów (image) oraz teleangiektazje na powiekach. W ciężkich postaciach mogą pojawiać się owrzodzenia.
Zajęcie mięśni Zajęcie mięśni poprzecznie prążkowanych występuje u 80–100% pacjentów (nieobecne w postaci amyopatycznej JDM). Typowym objawem jest symetryczne osłabienie mięśni proksymalnych kończyn górnych i dolnych, zginaczy szyi oraz mięśni brzucha. Często występują trudności przy wstawaniu z pozycji siedzącej (objaw Gowersa), zmęczenie i niezdarność ruchowa.
Zajęcie innych narządów
- Stawy: Symetryczne zapalenie wielostawowe (u 6–42% pacjentów), poranna sztywność, przykurcze.
- Zwapnienia dystroficzne: Odkładanie się wapnia w tkance podskórnej i powięziach, często związane z obecnością autoprzeciwciał anti-NXP2. (image)
- Przewód pokarmowy: Ryzyko niedokrwienia ściany jelita i perforacji, owrzodzenia błony śluzowej jamy ustnej.
- Płuca: Śródmiąższowe zapalenie płuc z ryzykiem rozwoju nadciśnienia płucnego.
- Miokardium: Zajęcie serca jest rzadkie u dzieci.
Rozpoznanie
Rozpoznanie JDM opiera się na objawach klinicznych i laboratoryjnych. Nowe kryteria klasyfikacyjne (Kryteria klasyfikacyjne dla idiopatycznych zapalnych miopatii: EULAR/ACR, 2017) dzielą pacjentów na trzy grupy według prawdopodobieństwa wystąpienia choroby. Wynik można łatwo obliczyć za pomocą internetowego kalkulatora: IIM calculator – English version.
Badania laboratoryjne
- Aktywność zapalna: Zazwyczaj nie jest wyraźnie podwyższona – OB i CRP mogą być prawidłowe lub jedynie nieznacznie zwiększone.
- Morfologia krwi: Zwykle w granicach normy, jednak może występować leukopenia, niedokrwistość Coombs-dodatnia lub trombocytopenia (szczególnie w zespole nakładania z toczniem rumieniowatym układowym).
- Immunofenotypowanie: Zwiększona liczba limfocytów B CD20+.
- Biochemia: Podwyższone enzymy mięśniowe (ALT, AST, LD, CK), zwiększone stężenie mioglobiny, podwyższony czynnik von Willebranda (marker waskulopatii).
- Immunologia: Dodatnie przeciwciała przeciwjądrowe (ANA), przeciwciała swoiste dla zapalenia mięśni (anti-Jo1, anti-Mi2, anti-SRP, anti-NXP2, anti-MDA5) oraz przeciwciała związane z zapaleniem mięśni (anti-dsDNA, anti-Sm, anti-SSA, anti-SSB, anti-U1-RNP). Niektóre przeciwciała korelują z określonymi objawami klinicznymi, takimi jak zajęcie płuc lub kalcynoza.
Badania obrazowe i czynnościowe
- Ocena siły mięśni: Z wykorzystaniem standaryzowanych testów, takich jak CMAS (Childhood Myositis Assessment Scale) i MMT8 (Manual Muscle Test).
- Rezonans magnetyczny (MRI) mięśni: Umożliwia uwidocznienie zmian zapalnych. MRI jest stosowany do monitorowania odpowiedzi na leczenie oraz do wyboru najbardziej odpowiedniego mięśnia do biopsji.
- Kapilaroskopia wałów paznokciowych: Ujawnia zmniejszenie liczby naczyń włosowatych, ich zmiany strukturalne i dezorganizację. Wynik koreluje z aktywnością choroby.
- Ocena kardiologiczna: EKG, echokardiografia i zdjęcie RTG klatki piersiowej mogą być pomocne w przypadku podejrzenia zajęcia serca lub płuc.
- Biopsja mięśnia: Wykonywana wybiórczo z mięśnia najbardziej zajętego według badania MRI; przydatna w określaniu rokowania.
- Badania czynnościowe płuc: Spirometria i pomiar zdolności dyfuzyjnej płuc pomagają wykryć możliwe powikłania oddechowe.
- Tomografia komputerowa płuc (HRCT): Umożliwia obiektywną ocenę obecności i rozległości śródmiąższowej choroby płuc.
Differencjalna diagnostyka
Ważne jest, aby odróżnić JDM od innych chorób o podobnych objawach:
- Choroby skóry: Łuszczyca (inna mikroangiopatia w badaniu wałów paznokciowych).
- Choroby nerwowo-mięśniowe: Dystrofia mięśniowa Duchenne’a i Beckera oraz inne dziedziczne miopatie, miopatie metaboliczne (choroba Pompego, zaburzenia β-oksydacji kwasów tłuszczowych).
- Choroby reumatologiczne: Młodzieńcze idiopatyczne zapalenie stawów, twardzina układowa, toczeń rumieniowaty układowy, mieszana choroba tkanki łącznej.
- Endokrynopatie: Niedoczynność tarczycy, nadczynność tarczycy.
- Zapalenie mięśni o etiologii infekcyjnej: Wirusy (grypa, coxsackie), bakteryjne zapalenie mięśni (pyomyositis), włośnica.
Sytuacje, w których należy rozważyć JDM
- U każdego dziecka z proksymalnym osłabieniem mięśni i minimalnymi zmianami skórnymi
- U dzieci z typowymi zmianami skórnymi, z osłabieniem mięśni lub bez niego
- U dzieci z podwyższonymi markerami uszkodzenia mięśni bez wyraźnego osłabienia i z minimalnymi lub brakiem zmian skórnych
Leczenie
Niefarmakologiczne
- Ochrona przed promieniowaniem UV.
- Rehabilitacja ukierunkowana na eliminację przykurczów i wzmocnienie mięśni.
- Zapobieganie aspiracji w zaburzeniach połykania.
Farmakologiczne
- Leczenie immunosupresyjne: Kortykosteroidy, metotreksat, cyklosporyna A
- Leczenie immunomodulujące: Dożylne immunoglobuliny (IVIG)
- Leczenie biologiczne: Zarezerwowane dla przypadków opornych (blokery TNF-α, przeciwciała anty-CD20).
Rokowanie
Dzięki wczesnemu i intensywnemu leczeniu immunosupresyjnemu śmiertelność JDM spadła do 1–2%. Niemniej jednak długoterminowa chorobowość pozostaje wysoka, a jedynie 30% pacjentów osiąga remisję bez potrzeby dalszej terapii. Częstość występowania kardiomiopatii i śródmiąższowej choroby płuc jest niższa niż u dorosłych pacjentów z dermatomiozytis. W przeciwieństwie do dorosłych, JDM nie jest związana z nowotworami. Wczesna diagnoza i szybkie rozpoczęcie leczenia mają kluczowe znaczenie dla rokowania pacjentów z JDM.
Tekst opracował Prokop Šeferna (prokop.seferna@vfn.cz), głównie na podstawie książki Dětská revmatologie v praxi, rozdziały 18 Idiopatické zápalové myopatie (Tomáš Dallos, dallos@nudch.eu) i 20 Juvenilní systémový lupus erythematosus (Hana Malcová – Hana.Malcova@fnmotol.cz, Veronika Vargová – veronika.vargova@upjs.sk).
Więcej postów dla pracowników służby zdrowia




